
RQM+ Medical Device and In Vitro Diagnostic Blog
Posts By: Stephen Biernacki
Sorry, no listings found for that Search. Try changing your fiter and search again.
-

Nancy Morrison's Keys to Thriving in Regulatory Affairs: Lessons Learned from a 30+ Year Career
on 11 April 2024 | By Stephen Biernacki, Marketing Principal
In this heartfelt and insightful video presentation, Nancy Morrison – as highly a respected professional as you'll ever find in regulatory affairs – shares her journey and the lessons she's learned along the way. As she embarks on her well-deserved retirement, Nancy generously...
Read More -

Join us at the 2021 RAPS U.S. Convergence
on 30 August 2021 | By Stephen Biernacki
It is that time of year again for RAPS Convergence and we cannot wait to join the largest annual gathering of regulatory professionals from around the world. The conference will be held virtually this year from 12-15 September 2021 and we will be joining in the fun with a Q&A...
Read More -

RAPS Webcast: PMCF User Feedback Surveys
on 8 April 2021 | By Stephen Biernacki
Update: the RAPS + RQM+ webcast is now available on demand here. Understand the ins and outs of PMCF user feedback surveys by registering for two upcoming and free RQM+ webinars: April 27 – PMCF User Feedback Surveys: Practical solutions for leveraging user feedback surveys as...
Read More -

PMCF User Feedback Surveys
on 17 February 2021 | By Stephen Biernacki
In this webinar we will provide practical solutions for leveraging user feedback surveys as PMCF activities under the EU MDR, presented by former notified body leadership and PMCF subject matter experts.
Read More -

Best Practices for Scientific Database Searching
on 18 January 2021 | By Stephen Biernacki
This on-demand webinar will address best practices in scientific database search techniques that can be applied in the performance of a clinical evaluation.
Read More -

Integrating Risk and Complaint Management
on 16 October 2020 | By Stephen Biernacki
Having strong integrated complaint and risk management processes is about more than just compliance. Watch this webinar and learn critical strategies and tactics for success. As with every R&Q webinar, all registrants will receive access to the slides and recording.
Read More -

Program Management for IVDR and EU MDR
on 8 September 2020 | By Stephen Biernacki
Whether you’re just starting to plan your IVDR program (or not sleeping well because you haven’t started yet) or well into EU MDR but ready to plan post-market surveillance activities, our September webinar will be helpful. All registrants will receive access to the slides and...
Read More -

Roundup: RQM+ at the 2020 RAPS Convergence
on 30 August 2020 | By Stephen Biernacki
RQM+ is counting down the days until we can join the RAPS community at the virtual RAPS Convergence 2020 on 13-16 September 2020. As many of you know, the Convergence is the largest annual gathering of regulatory professionals from around the world. While we'll miss interacting...
Read More -

Ranked: The most popular on-demand panel discussions from R&Q
on 24 August 2020 | By Stephen Biernacki
Note: A version of this post was initially published for the flagship online publication of RAPS, Regulatory Focus™ In an effort to further support the medical device industry during the COVID-19 pandemic, we've been spreading love more than ever in one of the best ways we know...
Read More -

Economic Operators: EU MDR and IVDR Requirements
on 3 August 2020 | By Stephen Biernacki
In our August 2020 webinar you'll learn about the EU MDR and IVDR requirements for economic operators.
Read More -
PMCF Plans
on 7 July 2020 | By Stephen Biernacki
In this on-demand webinar originally presented in July 2020, find out how to create detailed, compliant, and business-balanced PMCF plans.
Read More -

Case Studies of MDD and MDR Audit Findings... and Lessons Learned
on 8 May 2020 | By Stephen Biernacki
On-Demand - A webinar entirely dedicated to case studies.
Read More -

RQM+ Live! #5 — 5/14/20
on 1 May 2020 | By Stephen Biernacki
Chatting with Former FDA and Notified Body Representatives
Read More -
FDA Emergency Use Authorization (EUA) and EU MDR Article 59
on 27 April 2020 | By Stephen Biernacki
26 May 2020 - Processes, tips, and lessons learned.
Read More -

Join Us Weekly — Announcing DEVICE L❤️VE Live!
on 9 April 2020 | By Stephen Biernacki
The current pandemic is challenging for everyone to varying degrees. R&Q has the utmost respect, appreciation, and love for healthcare workers and all frontline workers across the globe. Our mission from the start has been to improve people's lives and our announcement today has...
Read More -
RAPS Webcast: PMS Requirements of the EU MDR
on 6 April 2020 | By Stephen Biernacki
Note: this webcast is now available on demand. R&Q is a Premium Solutions Partner of the Regulatory Affairs Professionals Society (RAPS) and on Wednesday, April 15th, we're joining forces to offer up a premium live webcast. Attendees will earn 1.5 RAC credits and the event is...
Read More -

Q&A: PMS Requirements of the EU MDR
on 3 April 2020 | By Stephen Biernacki
In February – just before COVID-19 began to seriously impact those of us in the United States – R&Q Executive Director of Regulatory & Quality Consulting Services, Nancy Morrison, presented the webinar PMS Requirements of the EU MDR: Implementation Challenges and Solutions. We...
Read More -

Structuring PERs under IVDR
on 1 April 2020 | By Stephen Biernacki
Whether you are interested in improved strategies for completing PERs or effectively completing your first PER, our April 2020 webinar can act as a critical guide.
Read More -

EU MDR Audits
on 5 March 2020 | By Stephen Biernacki
Preparing, managing and responding to nonconformances.
Read More -

Q&A: Developing an FDA Regulatory Strategy
on 28 February 2020 | By Stephen Biernacki
R&Q's first webinar of 2020 was the first in a two-part series on bringing your medical device to market in the United States. Part one addressed Class I and Class II devices, while part two (TBA in the spring) will cover Class III devices. The questions we received related to...
Read More -

PMS Requirements of the EU MDR
on 31 January 2020 | By Stephen Biernacki
Our February 2020 webinar will help you optimize every element of PMS/PMCF as it relates to the EU MDR at your organization.
Read More -

Developing an FDA Regulatory Strategy
on 8 January 2020 | By Stephen Biernacki
Our January 2020 webinar is designed to help you create a comprehensive regulatory strategy that enables you to successfully enter the US market.
Read More -

Announcing R&Q's new Vice President of Technical Services, Ralph Asencio
on 25 November 2019 | By Stephen Biernacki
Regulatory & Quality Solutions (R&Q) is absolutely delighted to announce Ralph Asencio as Vice President of Technical Services! R&Q has plenty to be thankful for in 2019. It's been an incredible year for a multitude of reasons, one of which has been the addition of key technical...
Read More -

Solving the EU MDR Labeling Puzzle
on 17 September 2019 | By Stephen Biernacki
Our December webinar will help you understand and act on requirements.
Read More -

Strategies for Successful IVDR Implementation
on 17 September 2019 | By Stephen Biernacki
Our November 2019 webinar looked at how to assess products and more.
Read More -

FDA Updates
on 17 September 2019 | By Stephen Biernacki
Our October 2019 webinar highlighted key areas of change related to the FDA.
Read More -

-

Q&A: Integrating CERs and Post-Market Surveillance
on 27 June 2019 | By Stephen Biernacki
While we're in the midst of taking the summer off from webinars (see you in the fall!) and excitedly preparing for our CER Virtual Workshop in September, we did have our most popular and best-reviewed webinar ever in May: Integrating CERs and Post-Market Surveillance. The...
Read More -

That's a wrap! A recap of our Advanced EU MDR and CER Workshops and what to do if you missed them.
on 9 April 2019 | By Stephen Biernacki
In March R&Q partnered with the Medical Alley Association in Minneapolis and MassMEDIC in Boston to offer industry-leading educational workshops on the EU MDR and CERs. R&Q recruited top industry experts, including representation from notified bodies BSI and GMED North America,...
Read More -

R&Q adds former TÜV Rheinland North America Lead Auditor of Medical to team
on 8 April 2019 | By Stephen Biernacki
Regulatory & Quality Solutions (R&Q) continues to grow aggressively – all while never sacrificing the high quality of service our clients have come to expect (this is a guiding principle and will never change) – and our latest addition to the team is particularly exciting. R&Q...
Read More -

EU MDR for Combination Products
on 25 March 2019 | By Stephen Biernacki
More companies will require notified body involvement.
Read More -

Top 10 EU MDR and CER Questions
on 25 February 2019 | By Stephen Biernacki
Answers to the most popular questions we receive.
Read More -

EU MDR / CER Portfolio Planning
on 25 February 2019 | By Stephen Biernacki
Know the essential EU MDR portfolio planning requirements.
Read More -

QMS for EU MDR
on 25 February 2019 | By Stephen Biernacki
Does your quality system meet the additional requirements?
Read More -

Risk Management Updates
on 25 February 2019 | By Stephen Biernacki
What to do with your process to meet the EU MDR/IVDR requirements.
Read More -
![🏅 Top 10 Questions: EU MDR and CER [Upcoming Webinar]](https://www.rqmplus.com/hs-fs/hubfs/RQ_Webinar_Promo_Top_10_1_21_19.png?length=450&name=RQ_Webinar_Promo_Top_10_1_21_19.png)
🏅 Top 10 Questions: EU MDR and CER [Upcoming Webinar]
on 25 January 2019 | By Stephen Biernacki
Are you registered for our next free webinar yet? It's one you won't want to miss and it's next week!
Read More -

Our Advanced EU MDR and CER Workshop is coming to California in December at DeviceTalks West
on 14 November 2018 | By Stephen Biernacki
Note: Presenters/Panelists listed below subject to change. R&Q is bringing our Advanced EU MDR and CER Workshop to DeviceTalks West! Join us Dec.12 for an extensive, detailed look at EU MDR and CERs through the lens of top industry experts who have successfully implemented...
Read More -

New webinars, major events... hello, fall!
on 11 September 2018 | By Stephen Biernacki
First thing's first: our free webinars are back. Over the next three months – beginning September 25 – we'll be covering combination products, EU MDR, and economic operators. Slides, recordings, and Q&A will be made available to everyone who registers. We've made it easy for you...
Read More -

Not your average EU MDR and CER workshop
on 20 July 2018 | By Stephen Biernacki
Note: Presenters/Panelists listed below subject to change. DeviceTalks Boston is Oct. 8-10 this year and as part of it, R&Q is helping to organize an Advanced EU MDR and CER Workshop. It's an extensive, detailed look at EU MDR and CERs through the lens of top industry experts...
Read More -

Taking it up a notch at DeviceTalks Minnesota
on 10 May 2018 | By Stephen Biernacki
DeviceTalks Minnesota is right around the corner and we couldn't possibly be more excited - really. June 4-5 is only a little over three weeks away! We've witnessed the growth of this event first-hand over the past few years and this year we're confident will be the best yet,...
Read More -

🔑 Unlock the Secrets to CERs in our May Webinar
on 7 May 2018 | By Stephen Biernacki
Unlock the secrets to complying with the increased requirements for CERs in May's free R&Q webinar: CERs – Tips, Tricks, and Lessons Learned The session will be on Tuesday, May 22 from 1:00pm - 2:00pm EST. The presentation slides, webinar recording, and Q&A will be made...
Read More -
![💼 EU MDR – Proactive Post-Market Surveillance [Upcoming Webinar]](https://www.rqmplus.com/hs-fs/hubfs/Webinars_2018/April%202018/RQ_Webinar_EUMDR_Proactive_Post-Market_Surveillance_Promo-min%20%281%29.png?length=450&name=RQ_Webinar_EUMDR_Proactive_Post-Market_Surveillance_Promo-min%20%281%29.png)
💼 EU MDR – Proactive Post-Market Surveillance [Upcoming Webinar]
on 20 April 2018 | By Stephen Biernacki
It's time for our next free R&Q Intelligence Series webinar. The session – EU MDR – Proactive Post-Market Surveillance: The requirements and staff it will take to do it. – will be held Tuesday, April 24ᵗʰ from 1:00pm - 2:00pm EST. If you can't view the webinar live, we encourage...
Read More -

Webinar Q&A: Preparing your CER for MDR
on 8 January 2018 | By Stephen Biernacki
Note: R&Q's first Intelligence Series webinar of 2018 will be held on January 23rd – QMS for EU MDR: Does your quality system meet the additional requirements? Sign up here! -- Back in November and as part of the R&Q Intelligence Series, we held the webinar, Preparing your CER...
Read More -

Welcome, Jacob Foster: Regulatory and Quality Solutions' New Senior Principal Engineer
on 14 November 2017 | By Stephen Biernacki
Jacob Foster brings 20+ years of medical device and pharmaceutical regulatory, quality, and project management experience to R&Q. He also brings an engaging personality, eager to do more for clients. Pittsburgh, PA -- Regulatory and Quality Solutions LLC (R&Q) exists to improve...
Read More -
![Preparing your CER for MDR [Upcoming Webinar]](https://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R%26Q%20Intelligence%20Series%202017/2017%20November%20-%20Preparing%20your%20CER%20for%20MDR/RQ_Webinar_Preparing_CER_for_MDR_Website_Promo_10_31_17-min.png?length=450&name=RQ_Webinar_Preparing_CER_for_MDR_Website_Promo_10_31_17-min.png)
Preparing your CER for MDR [Upcoming Webinar]
on 13 November 2017 | By Stephen Biernacki
The upcoming EU MDR requires enhanced clinical evidence to support the device whether you have Class I or Class III products. For products that have been on the market for a long time there may be limited or no clinical studies, and yet, the new European Medical Device...
Read More -

ISO 13485:2016 Companion Handbook Now Available
on 24 October 2017 | By Stephen Biernacki
ISO has published a companion handbook to ISO 13485:2016, Medical devices-Quality management systems - Requirements for regulatory purposes. From AAMI: It provides users with practical guidance and accurate interpretation of the requirements specified in the standard. It offers...
Read More -

Webinar Q&A: EU MDR / CER Portfolio Planning
on 2 October 2017 | By Stephen Biernacki
In September and as part of the R&Q Intelligence Series, we conducted the webinar, EU MDR / CER Portfolio Planning: Know the essential EU MDR portfolio planning requirements. Portfolio planning is a key aspect of interfacing with your notified body. The upcoming EU MDR changes...
Read More -

Webinar Q&A: Risk-Based Approach to ISO 13485:2016
on 19 September 2017 | By Stephen Biernacki
Last month and as part of the R&Q Intelligence Series we conducted the webinar, Risk-Based Approach to ISO 13485:2016: Risk considerations and implementation. Risk considerations and implementation of a risk-based approach in your QMS processes is one of the most significant...
Read More -
![EU MDR / CER Portfolio Planning: The Essential Requirements [Free Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/September%20-%20Portfolio%20Planning/Webinar_Promo_September_Portfolio_Planning_Website-min.png?length=450&name=Webinar_Promo_September_Portfolio_Planning_Website-min.png)
EU MDR / CER Portfolio Planning: The Essential Requirements [Free Webinar]
on 12 September 2017 | By Stephen Biernacki
What? Please join us for September's free R&Q Intelligence Series webinar: EU MDR / CER Portfolio Planning: Know the essential EU MDR portfolio planning requirements. The session will take place Tuesday, September 26 from 1:00pm - 2:00pm EST. Portfolio planning is a key aspect...
Read More -

MEDDEV 2.7/1 & CERs: Questions and Answers
on 21 August 2017 | By Stephen Biernacki
Clinical evaluation requirements have been changing, with the latest impact coming from MEDDEV 2.7/1 Rev 4. Preparing for and meeting these requirements is important because the grace period offered by some notified bodies is ending and clinical evaluation reports (CERs) are...
Read More -

Clarifying the Clinical Evaluation Requirements: A Case Study
on 1 August 2017 | By Stephen Biernacki
About R&Q's Case Studies: We hope this Clinical Evaluation Report case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies and other resources are available. Questions on this...
Read More -

EU MDR: Your Questions, Our Answers
on 27 July 2017 | By Stephen Biernacki
On May 5, 2017 the EU MDR (Medical Device Regulation) was published in the EU Official Journal. This date starts the three-year mandatory transition period where the MDR will replace the MDD (Medical Device Directive). The expanded scope of the MDR combined with the time frames...
Read More -

Regulatory and Quality Solutions Names Julie Maes Director of Territory Operations – Northern Lakes Region
on 24 July 2017 | By Stephen Biernacki
Maes brings 25+ years of medical device regulatory, quality, and project management experience to R&Q. Pittsburgh, PA -- Regulatory and Quality Solutions LLC (R&Q), a provider of industry-leading regulatory and quality engineering services to medical device and combination...
Read More -
![[Free R&Q Webinar] Cybersecurity for Medical Devices: The regulatory and quality ramifications.](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/July%20-%20Cybersecurity/Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png?length=450&name=Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png)
[Free R&Q Webinar] Cybersecurity for Medical Devices: The regulatory and quality ramifications.
on 6 July 2017 | By Stephen Biernacki
What? Please join us for a free R&Q Intelligence Series webinar: Cybersecurity for Medical Devices: The regulatory and quality ramifications. The session will be held on Tuesday, July 25 from 1:00pm - 2:00pm EST.
Read More -
![MEDDEV 2.7/1 & CERs: Know the Changes and What to Do [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/May%20-%20MEDDEV/Webinar_Promo_May_MEDDEV_v2-min.png?length=450&name=Webinar_Promo_May_MEDDEV_v2-min.png)
MEDDEV 2.7/1 & CERs: Know the Changes and What to Do [Webinar]
on 2 May 2017 | By Stephen Biernacki
Part of your month, [almost*] every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and...
Read More -
![EU MDR: Assessing the Impact and Next Steps [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/April%20-%20EU%20MDR/Webinar_Promo_April_EU_MDR_v2-min.png?length=450&name=Webinar_Promo_April_EU_MDR_v2-min.png)
EU MDR: Assessing the Impact and Next Steps [Webinar]
on 5 April 2017 | By Stephen Biernacki
Part of your month, every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and topics in...
Read More -
![MDSAP: What is it? Is it for me? How will I get it done? [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/March%20-%20MDSAP/Webinar_Promo_March-min.png?length=450&name=Webinar_Promo_March-min.png)
MDSAP: What is it? Is it for me? How will I get it done? [Webinar]
on 7 March 2017 | By Stephen Biernacki
Part of your month, every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and topics in...
Read More -
![A Risk-Based Approach to Your QMS Implementation - ISO 13485:2016 and ISO 9001:2015 [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Headshots/Optimized_for_Web/mark_swanson-min.jpg?length=450&name=mark_swanson-min.jpg)
A Risk-Based Approach to Your QMS Implementation - ISO 13485:2016 and ISO 9001:2015 [Webinar]
on 12 February 2017 | By Stephen Biernacki
Part of your month, every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and topics in...
Read More -

Acquisition Regulatory Assessments: A Case Study
on 25 January 2017 | By Stephen Biernacki
About R&Q's Case Studies: We hope this Acquisition Regulatory Assessments case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies and other resources are available. Questions on this...
Read More -

Regulatory and Quality Solutions (R&Q) Certified By the Women’s Business Enterprise National Council
on 9 January 2017 | By Stephen Biernacki
R&Q, provider of industry-leading regulatory and quality consulting and engineering services to medical device companies, is proud to announce national certification as a Women’s Business Enterprise by the Women's Business Enterprise Council-PA-DE-sNJ, a regional certifying...
Read More -

Headway in PA: Announcing Dedicated Operations Directors in Pittsburgh and Philadelphia
on 16 December 2016 | By Stephen Biernacki
In 2016 R&Q expanded its activities and the amount of clients served in the northeast (specifically, the greater Boston region), formally launched R&Q in Minnesota, and continued to service new and existing clients throughout the state of Ohio. In 2017 we plan to strengthen our...
Read More -

Process Validation Case Study: Know What You Know... and What You Don't
on 13 December 2016 | By Stephen Biernacki
About R&Q case studies: We hope this process validation case study helps you understand how we serve our medical device clients in this area. If you have specific process validation questions please contact us and we'll be happy to help. Looking to solve a different problem?...
Read More -

Vital To-Do for 2017: Prepare for ISO 13485:2016
on 7 November 2016 | By Stephen Biernacki
“Procrastination is the bad habit of putting of until the day after tomorrow what should have been done the day before yesterday.” - Napolean Hill It's really never too early, especially when it comes to renewing the certifications you need to keep your product on the market. In...
Read More -

Slides and Webinar Recording: Chemical Characterization Requirements
on 28 October 2016 | By Stephen Biernacki
Missed our webinar with iuvo BioScience - Chemical Characterization Requirements: ISO 10993-18 and Extractables & Leachables Requirements? Good news: you can now download the slides and view the recording.
Read More -

Next Up: FDA Regulatory 101 with Combination Products Spotlight at Heal Ohio Conference
on 19 October 2016 | By Stephen Biernacki
This is the last FDA Regulatory 101 installment of the year for our partnership with BioOhio. After events in Cincinnati and Columbus, the October 27th event will be held during the 2016 Heal Ohio Conference at the University of Akron. Former FDA Consumer Safety Officer Jake...
Read More -

CAPA on CAPA: Webinar Slides and Recording
on 7 October 2016 | By Stephen Biernacki
Missed our CAPA on CAPA webinar with MassMEDIC? Now you can download the slides and view the recording.
Read More -

Free Webinar: Chemical Characterization Requirements: ISO 10993-18 and Extractables & Leachables Requirements
on 28 September 2016 | By Stephen Biernacki
What? This joint webinar presented by Regulatory and Quality Solutions (R&Q) and iuvo BioScience is focused on the requirements for chemical characterization in the healthcare industry. Specifically, it will discuss the standards required for ISO 10993-18 chemical...
Read More -

Minnesota Momentum: R&Q Hires Member of ISO Technical Committee 210, Mark Swanson
on 21 September 2016 | By Stephen Biernacki
Mark Swanson is R&Q's new Director of Minneapolis–Saint Paul (MSP) Territory. Welcome, Mark! Mark's addition firmly establishes R&Q in the Twin Cities and prepares R&Q to handle growth in the region. The hiring also builds upon R&Q's existing technical leadership, increasing...
Read More -

Revamping Supplier Quality: A Case Study
on 12 September 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope this Supplier Quality Remediation case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies are available. Questions on supplier quality remediation?...
Read More -

When CAPA Needs a Corrective Action: A CAPA Remediation Case Study
on 8 September 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope this case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies are available. Questions? Contact us. Challenge The FDA found problems you didn't think...
Read More -

Med Device and Med Tech Startups: Master Regulatory Basics and More! September 22 in Philly
on 1 September 2016 | By Stephen Biernacki
We all gotta start somewhere... and Pennsylvania Bio and Quorum have helped us put together an invaluable event to help your early-stage medical device company hit the ground running. Seasoned experts, such as R&Q's President Maria Fagan and Buchanan Ingersoll & Rooney's Will...
Read More -

Free Webinar: CAPA on CAPA, Presented by MassMEDIC and R&Q - Register Now!
on 16 August 2016 | By Stephen Biernacki
CAPA is often overlooked as an integral component of a quality system. More often than not, the FDA can trace deficiencies in a system directly back to the CAPA system. However, an effective quality system needs more than just a rework of the outcome of the ineffective CAPA...
Read More -

R&Q's Christine Santagate Featured in Medical Design & Outsourcing
on 3 August 2016 | By Stephen Biernacki
Staying at the forefront of the medical device industry takes work - a lot of it, in fact. Communication with R&Q's peers in the industry is in large part how our employees do it, and a big part of that is addressing hot topics in the industry. R&Q's own Client Solutions Advisor...
Read More -

July is a BIG month for R&Q in Columbus, OH
on 12 July 2016 | By Stephen Biernacki
■ ■ ■ Are you an Ohio Bioscience Expo & Showcase attendee here to register for the FDA Regulatory 101 Series? ■ ■ ■ The Ohio Bioscience Expo & Showcase and next FDA Regulatory 101 Series event are on July 27th and 28th, respectively. If you're outside of Columbus but in Ohio,...
Read More -

10 Tips for Adopting the Updated ISO 13485:2016 Standards: Webinar Slides and Recording
on 7 July 2016 | By Stephen Biernacki
Missed our latest webinar on this year's ISO 13485 changes? Now you can download the slides and recording. R&Q's Director of Regulatory Affairs Nancy Morrison and Client Solutions Advisor Christine Santagate walked through 10 tips for adopting the latest changes. From conducting...
Read More -

FDA Approved: A Human Factors and Usability Case Study
on 22 June 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope you enjoy our latest case study. View all of our available Case Studies. Subscribing to our blog is the best way to know when future case studies are available. FDA Approved: Creating and Implementing a Compliant Usability Engineering Process...
Read More -

4 Things R&Q Learned At MD&M East
on 21 June 2016 | By Stephen Biernacki
What an action-packed three days MD&M East was this year! Our team is exhausted, but in the absolute best way possible. I'm not sure how anyone who attended the show could've escaped the exhaustion... as the exhibit floor was BIG. If you're reading this and attended, hopefullly...
Read More -

R&Q: Engineered by Women
on 24 May 2016 | By Stephen Biernacki
Did you know that women are half as likely as men to start businesses? That's a statistic R&Q wants to help change. Aside from the importance of gender and fairness issues, women entrepreneurship fuels economic growth. R&Q is Engineered by Women. We're proud to have been started...
Read More -
A Whole New World of UDI: Slides and Recording
on 20 May 2016 | By Stephen Biernacki
If you have any UDI needs or questions, do not miss this post.
Read More -

Presentation and Networking at PA Bio: Practical Considerations for Avoiding FDA Compliance Problems (BEING RESCHEDULED)
on 11 May 2016 | By Stephen Biernacki
NOTE: This event has been cancelled and is being rescheduled. Organizer Regulatory and Quality Solutions (R&Q) What? A presentation and networking event over lunch at PA Bio: Practical Considerations for Avoiding FDA Compliance Problems, and How to Manage Them If They Arise In...
Read More -

The New ISO 13485 Standards are Here! Sign Up for a Free R&Q Webinar
on 11 May 2016 | By Stephen Biernacki
The world's most well known standards for medical device quality management have been updated for the first time since 2003. Released in March by the International Organization for Standardization, ISO 13485 provides the universal requirements for manufacturers and service...
Read More -
MassMEDIC Webinar: A Whole New World of UDI - May 18
on 28 April 2016 | By Stephen Biernacki
If you missed R&Q's Paul Robinson discuss UDI during R&Q's afternoon workshop at BIOMEDevice in April, here's a second chance. A similar presentation and discussion will be offered in conjunction with MassMEDIC on Wednesday, May 18th at 11:30am EST. All the details - including...
Read More -

AdvaMed Webinar: Avoiding and Managing FDA Compliance Problems - Monday, May 9th
on 15 April 2016 | By Stephen Biernacki
Did you miss former FDAer Jake O'Donnell speak about avoiding and managing FDA compliance problems during R&Q's afternoon workshop at BIOMEDevice earlier this week? If so, we have news especially for you: he will be presenting on a similar topic via a webinar on Monday, May 9th...
Read More -

NEW White Paper - Practical Considerations for Avoiding Regulatory Escalation: R&Q's Latest, In-Depth White Paper from a Former FDA Consumer Safety Officer
on 13 April 2016 | By Stephen Biernacki
There’s a formula for effectively managing an adverse FDA inspection and the newest R&Q white paper shares it. Learn all the practical considerations for avoiding regulatory escalation from a former FDA Consumer Safety Officer: R&Q's Jake O'Donnell. This is your free, ultimate...
Read More -

Minnesota, Here We Come: See You at the 2016 Medical Alley Annual Meeting, April 27th
on 23 March 2016 | By Stephen Biernacki
R&Q will be a Supporting Sponsor at the Medical Alley Association's 2016 Annual Meeting, April 27th. If you're not familiar, the association is a state-based member organization servicing the health technology community who works to promote Minnesota's Medical Alley by...
Read More -

R&Q's Can't-Miss Education Event of the Year: An Afternoon Training Workshop at BIOMEDevice in Boston, April 13th
on 8 March 2016 | By Stephen Biernacki
Those in the northeast, get ready! Not only will R&Q be exhibiting at this year's BIOMEDevice show (booth 640) and sponsoring MassMEDIC's Annual Conference, but we'll be presenting an afternoon training workshop (three separate sessions) free to attendees of the BIOMEDevice show...
Read More -

When Benefit Outweighs Risk: Creating a Successful Clinical Evaluation Report - A Case Study
on 18 February 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope you enjoy our third case study. View all of our available Case Studies. Subscribing to our blog is the best way to know when future case studies are available. Challenge High volume of data... and differing opinions. A client's product is...
Read More -

5 Heads are Better Than 1: De-Risking Your Regulatory Pathway Using a Team Approach - A Case Study
on 15 February 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope you find value in our second case study. View all of our available Case Studies. Subscribing to our blog is the best way to know when future case studies are available. Challenge Regulation doesn't have to be a barrier to innovation. A client...
Read More -

Introducing Our First In A Series of New RQM+ Case Studies: Design History File Remediation
on 12 February 2016 | By Stephen Biernacki
About RQM+ Case Studies: Our goal in producing case studies is to succinctly demonstrate how RQM+ applied our unique expertise to tackle a client's challenge, implement a competent solution, and demonstrate real results. We will post each case study to our blog and offer a...
Read More -

Announcing the 2016 FDA Regulatory 101 Series
on 8 February 2016 | By Stephen Biernacki
Medical device professionals in Ohio, are you ready? Last year R&Q teamed with BioOhio to present an ongoing series of 101 medical device events throughout the year. Thanks to participation and feedback from ambitious medical device professionals across Ohio - and the support of...
Read More -

Hear R&Q and Other Industry Experts Discuss 510(k) Submissions at AdvaMed's Workshop In Irvine, CA
on 2 February 2016 | By Stephen Biernacki
Join R&Q's Marilyn Waxberg and Nancy Morrison as they speak along with other industry experts at AdvaMed's 510(k) Submissions Strategy Workshop in Irvine, CA this month. The event is scheduled for two days: February 22-23. Marilyn is R&Q's Senior Principal Advisor and Nancy is...
Read More -
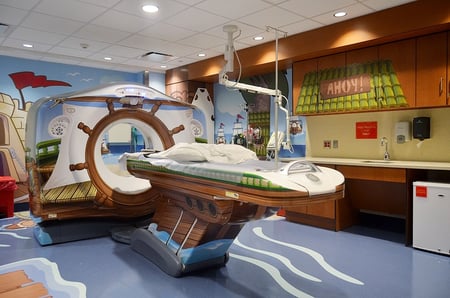
Revisiting a Classic: The Pirate-Themed CT Scanner
on 26 January 2016 | By Stephen Biernacki
Behold medical device ingenuity at its finest. In 2013 the NewYork-Presbyterian Morgan Stanley Children's Hospital purchased a pirate-themed CT scanner, and the result is an incredible way to put children at ease before being scanned. See the rest of the creativity in pictures...
Read More -

R&Q Is Sponsoring The M2D2 $100K Challenge and A Summary of Important Dates
on 22 January 2016 | By Stephen Biernacki
The University of Massachusetts Medical Device Development Center (M2D2) has announced the M2D2 $100K Challenge 2016, a nationwide competition that showcases innovative ideas of early-stage medical device, diagnostic, and biotech companies. R&Q is proud to announce that we are...
Read More -

Presentation and Networking: Is A Medical Device Hiding In Your Product Portfolio? Join R&Q at MassBio!
on 14 January 2016 | By Stephen Biernacki
Organizer Us! Regulatory and Quality Solutions (R&Q) What? A presentation and networking event over lunch addressing how an accessory or tool, in reality, is often a medical device. This session will discuss combination products and the use of accessories with drugs and...
Read More -

Announcing R&Q's Newest Senior FDA Compliance Principal: Former FDA Consumer Safety Officer Jake O'Donnell
on 6 January 2016 | By Stephen Biernacki
R&Q is thrilled to announce the newest member of our team. Jake O'Donnell will act as Senior FDA Compliance Principal beginning February 2016. Jake formerly acted as Consumer Safety Officer (CSO) at the FDA for 20 years.
Read More -
We Made a Video About R&Q! Here's Why.
on 1 December 2015 | By Stephen Biernacki
One of R&Q's priorities in 2015 was to find new, distinctive ways to tell our story. We wanted to make it abundantly clear to potential clients and employees that R&Q is first and foremost about improving lives. We also wanted to clearly explain how we improve lives. The result...
Read More -

Welcome To R&Q's New Website!
on 13 November 2015 | By Stephen Biernacki
Welcome to R&Q's updated website! To state it plainly, we've revamped our website to better serve the needs of regulatory and quality professionals in the medical device industry. We hope you find it easy to use. Some changes include more clearly outlined services, additional...
Read More -
510(k)-Exempt Devices
on 5 September 2013 | By Stephen Biernacki
In the FDA-regulated medical device world, there are 3 classifications for medical devices: Class I, Class II and Class III. The FDA (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/default.htm) provides a rather in-depth overview of how...
Read More


![FDA Inspections Summit – EU MDR: The Final Push [Panel]](https://www.rqmplus.com/hs-fs/hubfs/Events/2019/RAPS%20Regulatory%20Convergence/IMG_3664-min.jpeg?length=450&name=IMG_3664-min.jpeg)


















![EU MDR / CER Portfolio Planning: The Essential Requirements [Free Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/September%20-%20Portfolio%20Planning/Webinar_Promo_September_Portfolio_Planning_Website-min.png?length=450&name=Webinar_Promo_September_Portfolio_Planning_Website-min.png)




![[Free R&Q Webinar] Cybersecurity for Medical Devices: The regulatory and quality ramifications.](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/July%20-%20Cybersecurity/Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png?length=450&name=Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png)
![MEDDEV 2.7/1 & CERs: Know the Changes and What to Do [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/May%20-%20MEDDEV/Webinar_Promo_May_MEDDEV_v2-min.png?length=450&name=Webinar_Promo_May_MEDDEV_v2-min.png)
![EU MDR: Assessing the Impact and Next Steps [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/April%20-%20EU%20MDR/Webinar_Promo_April_EU_MDR_v2-min.png?length=450&name=Webinar_Promo_April_EU_MDR_v2-min.png)
![MDSAP: What is it? Is it for me? How will I get it done? [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/March%20-%20MDSAP/Webinar_Promo_March-min.png?length=450&name=Webinar_Promo_March-min.png)
![A Risk-Based Approach to Your QMS Implementation - ISO 13485:2016 and ISO 9001:2015 [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Headshots/Optimized_for_Web/mark_swanson-min.jpg?length=450&name=mark_swanson-min.jpg)





























We are passionate about your success. Tell us more about your regulatory and quality needs to learn about how we can help.

GLOBAL BOTTOM CTA INSTRUCTIONS:
To display custom copy instead of global copy in this section, please go to Show Global Content for Bottom CTA? toggle in the "Contents" tab to the left, toggle it off, save, and then REFRESH the page editor, the custom text will then show up and ready to be edited.
Turning the global content back on will be the same process, go to the toggle and toggle it back on, save and refresh!
.svg "white RQM logo")




