
How RQM+ Adds Value
-

Clinical and Regulatory Expertise
Our powerhouse team includes former notified body leaders, regulatory experts, medical doctors, clinicians, statisticians, and device technology experts that cover every type of medical device.
The team is led by Dr. Amie Smirthwaite, recent Global Head of Clinical Compliance at BSI, who was a member of multiple ISO technical committees and provided significant contributions to the European Commission Clinical Investigations and Evaluations Expert Group, including subgroups which authored multiple MDCG guidance documents on clinical evaluation and PMCF topics.
Our team members understand the intent behind the regulation and what notified body reviewers expect to see. With RQM+ as your strategic partner, you can expect see fewer notified body questions and faster time to certification.
-

Proven Processes
Our objective is always to provide a complete and cohesive story of sufficient clinical evidence in your technical documentation. The CER, PMS, PMCF, risk file, and IFU must all be singing in harmony, providing the notified body reviewer with confidence in your evidence and path forward. Our finely honed processes and tools ensure clarity and consistency across all documentation. We also use multiple checkpoints to ensure alignment and quality control checks to verify accuracy.
-

Advanced Methodologies
We create a clear mapping between the intended use and the evidence for demonstration of clinical conformity via a clinical evidence matrix. This is the single most effective tool for not only justifying that you have sufficient clinical evidence, but also the adequacy of your PMS/PMCF plans.
We streamline the gap analysis process so we can efficiently identify areas of weakness in clinical evidence across product claims and indications and create a plan for filling the gaps. We also employ project management tools and software such as Distiller, Embase, and a range of platforms to perform thorough literature searches and associated tasks to create comprehensive CERs.
Clinical Roadmap
Our experts will help you develop a clinical evaluation strategy to achieve your business objectives and notified body approval.
We provide innovative strategies for using existing data along with your PMCF plan based upon our experience with a wide range of notified bodies.
From CEPs to SSCPs, we enable you to tell a compelling story, making your documentation appealing to regulators.
Our integrated approach reduces time to certification with fewer notified body questions and nonconformities.



Quality System Solutions
CERs cannot be created in a bubble by the clinical team. There are inputs and outputs between clinical, post-market surveillance, and risk management. Integrating the processes optimizes resources and reduces the risk of audit findings. Our experts will help you update and revise your quality system so that all your processes are working together efficiently. This will ensure that you can meet the requirements of the MDR now and in the future.

NB Response Support
The notified body may not interpret the MDR's requirements in the same way you do and you only have three rounds of Q&A to gain alignment and approval. Additionally, a deficiency that calls for more clinical data could result in an expensive study or the loss of product certification. With so much on the line, a different perspective and proposed response plan from our former notified body leaders and subject matter experts could make all the difference.


CER Remediation
The RQM+ team will assess your CER process and documentation by product line, providing you with a detailed gap analysis and remediation plan. Our team of experts can effectively execute a remediation of any size, minimizing the burden on your team so you can maintain focus on new products.
Our CER project managers act as the single point of contact for all client needs and are responsible for leading meetings, developing schedules for deliverables, monitoring progress, and driving results. This includes actively managing the client approval process and coordinating independent review of all documentation.
For ongoing compliance, we ensure client documentation is updated as new data becomes available and then provided to notified bodies by the required deadlines.

Featured Leader

Amie Smirthwaite, Ph.D.
Senior VP, Intelligence & Innovation
Dr. Amie Smirthwaite joined RQM+ in March of 2020. A clinical and regulatory affairs expert, Amie has over 25 years of postdoctoral experience in medical devices, spanning new product development, quality and regulatory systems, and clinical data evaluation. She is leading the RQM+ Intelligence and Innovation team and brings a wealth of knowledge and experience having worked for medical device companies, academic institutions, and BSI.Entrusting your CERs to a consulting firm requires confidence in the team. With RQM+, you get a level of clinical and regulatory expertise and client dedication you won’t find anywhere else.

Tap into the RQM+ Knowledge Center
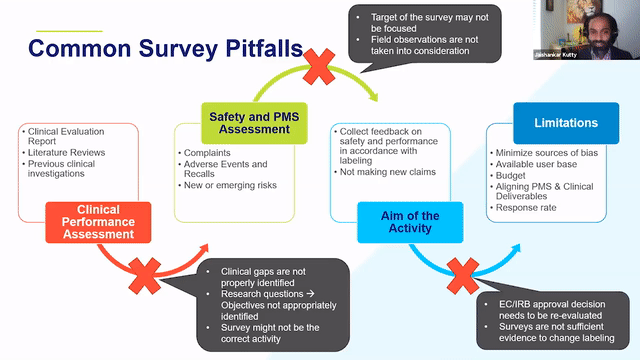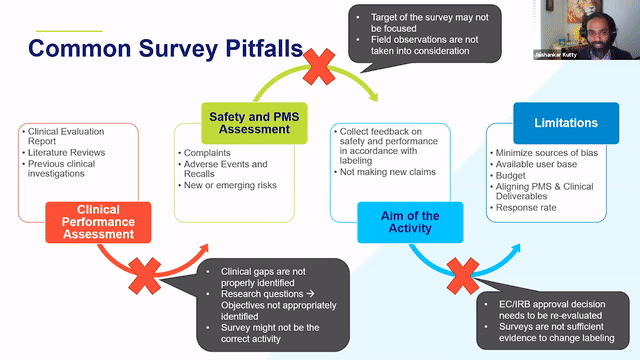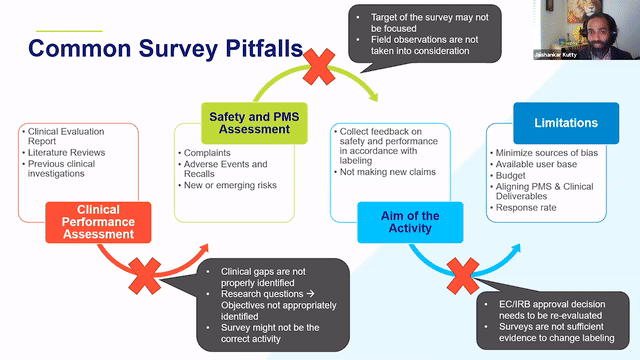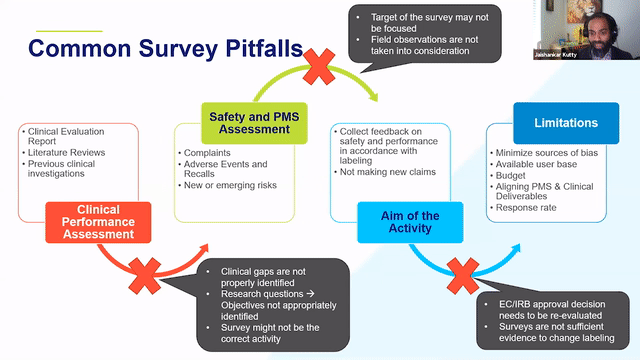
RAPS + RQM+ Webcast —PMCF User Feedback Surveys

How to Create a Compliant Periodic Safety Update Report (PSUR) Under EU MDR and EU IVDR

PMCF Under MDR
The integration of clinical, regulatory, risk management and post market surveillance is a difficult and confusing task. How should the data flow and where does it start?

Best Practices in Scientific Database Searching

Best Practices for Integrating CERs and PMS Under MDR
We are passionate about your success. Tell us more about your needs so we can help.

GLOBAL BOTTOM CTA INSTRUCTIONS:
To display custom copy instead of global copy in this section, please go to Show Global Content for Bottom CTA? toggle in the "Contents" tab to the left, toggle it off, save, and then REFRESH the page editor, the custom text will then show up and ready to be edited.
Turning the global content back on will be the same process, go to the toggle and toggle it back on, save and refresh!
.svg "white RQM logo")




